In March 2010, scientists announced the discovery of a finger bone fragment of a juvenile female that lived about 41,000 years ago, found in Denisova Cave in Altai Krai, Russia. The full genome of this
Denisova hominin was recently
made available. The
Unified genotyper was used to to add dbSNP 132 rs#s to the genome. The
resulting VCF file can be read by Promethease to produce:
http://files.snpedia.com/reports/promethease_data/genome_DenisovaPinky_ui2.html
Through the 5519 SNPedia annotated snps we can learn more about this distant relative.
APOE
As first
noted by John Hawks the snp
rs7412 couldn't be reliably called, but
rs429358 and
rs4420638 were. These are consistent with an E4/E4.
At present, the frequency of APOE*4 within all the major human groups remains higher in those populations…where an economy of foraging still exists, or food supply is now or has until recently been scarce, sporadically available or qualitatively poor. Under these environmental conditions, carrying the APOE*4 could be still useful.
--source PMID 10738542.
Hair Morphology
rs3124314(C;C) suggest some curliness to the hair, while
rs261360(A;G) is notable for heterozygosity. 5 other snps (rs12623288(A;A), rs1268789(G;G), rs1454292(T;T), rs6732426(T;T), rs908922(A;A)) are all consistent with straighter hair.
Skin color
Sex
This should be female, but 23508 rs#s on the Y-chrom were found, with only
rs9786465 being in SNPedia. The Unified Genotyper provides a partial explanation about the challenges of
calling sex chromosomes.
Caveat lector
The traits below are more about notoriously difficult to define and phenotype even in modern humans, and in a single sample of 40k year old non-human dna they should be covered in NaCl. However, deeper analysis needs to begin somewhere so ...
Optimism
rs53576(G;G) in the Oxytocin receptor (OXTR). This genotype appears to be significantly better at accurately reading the emotions of others by observing their faces than were the remaining three-quarters of subjects, with (A;A) or (A;G). (G;G) individuals were also less likely to startle when blasted by a loud noise, or to become stressed at the prospect of such a noise.
Intelligence
Quite a few rare genos for intelligence with a possible emphasis on spatial working memory.
Speech
rs2710102 in CNTNAP2 has been associated with impaired speech development.
Methods
Denisova data lives in the eu-west-1b region of the Amazon cloud as snap-3cc2de54.
Processed with
GenomeAnalysisTK-1.4-25-g23e7f1b
dbsnp acquired from
ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/1.2/b37/dbsnp_132.b37.vcf.gz
Generated via
java -jar GenomeAnalysisTK.jar -R /mnt/mydata/human_g1k_v37.fasta -T UnifiedGenotyper -I /mnt/den/denisova_genome/T_hg19_1000g.bam --dbsnp /mnt/mydata/dbsnp_132.b37.vcf -o /mnt/mydata/snps.raw.vcf > /mnt/mydata/alog.txt
Log finishes with
INFO 10:11:25,697 UnifiedGenotyper - Visited bases 3101804739
INFO 10:11:25,697 UnifiedGenotyper - Callable bases 2862033547
INFO 10:11:25,698 UnifiedGenotyper - Confidently called bases 112644898
INFO 10:11:25,698 UnifiedGenotyper - % callable bases of all loci 92.270
INFO 10:11:25,698 UnifiedGenotyper - % confidently called bases of all loci 3.632
INFO 10:11:25,698 UnifiedGenotyper - % confidently called bases of callable loci 3.936
INFO 10:11:25,699 UnifiedGenotyper - Actual calls made 4989617
INFO 10:11:25,714 TraversalEngine - Total runtime 66799.27 secs, 1113.32 min, 18.56 hours
INFO 10:11:25,823 TraversalEngine - 0 reads were filtered out during traversal out of 1424486071 total (0.00%)
It was run on an m1.large, but never managed to use both cpus, instead maxing out at 50% cpu usage.
[ec2-user@ip-10-234-51-252 den]$ df -H
Filesystem Size Used Avail Use% Mounted on
/dev/xvda1 8.5G 2.0G 6.5G 24% /
tmpfs 4.0G 0 4.0G 0% /dev/shm
/dev/xvdf 212G 170G 31G 85% /mnt/den
/dev/xvdg 159G 5.8G 145G 4% /mnt/mydata
[ec2-user@ip-10-234-51-252 mydata]$ ls -tral
total 5416212
drwxr-xr-x 4 root root 4096 Feb 9 15:03 ..
drwx------ 2 root root 16384 Feb 9 15:06 lost+found
-rw-rw-r-- 1 ec2-user ec2-user 4578627636 Feb 9 15:35 dbsnp_132.b37.vcf
-rw-rw-r-- 1 ec2-user ec2-user 12379076 Feb 9 15:35 dbsnp_132.b37.vcf.idx
-rw-rw-r-- 1 ec2-user ec2-user 942611555 Feb 10 10:11 snps.raw.vcf
-rw-rw-r-- 1 ec2-user ec2-user 12394492 Feb 10 10:13 snps.raw.vcf.idx
-rw-rw-r-- 1 ec2-user ec2-user 142784 Feb 10 10:13 alog.txt
drwxrwxrwx 3 root root 4096 Feb 10 12:16 .
Half of the /mnt/den is the chimpanzee data, so the alignment to human was 85gb for the T_hg19_1000g.bam + 9mb for the index.
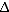 32 homozygous individuals, makes it unlikely that allogeneic HSC transplants using cells from ccr5
32 homozygous individuals, makes it unlikely that allogeneic HSC transplants using cells from ccr5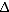 32 homozygous donors will become a widespread treatment option, and has prompted attempts to mimic the genetic knockout”.(PMID 22470838)
32 homozygous donors will become a widespread treatment option, and has prompted attempts to mimic the genetic knockout”.(PMID 22470838)