Timothy Ray Brown was suffering from both HIV and leukemia, and during the course of treatment for his leukemia, researchers in Berlin realized there might be a way to take advantage of rs333’s ability to resist HIV. After chemotherapy and radiation treatments, the researchers transplanted bone marrow from a donor with 2 copies of the rs333 deletion. These donor cells were able to reconstitute his blood.
The transplant took place in 2007, and doctors waited and watched for years while his body cleaned out the HIV and repaired itself. Finally publishing in 2010 and 2011, they were able to conclude that the 'SNP transplant' cured their patient’s HIV PMID 21148083 “Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation”.
Now in 2012 a Phase 2 clinical trial has begun recruiting patients. The protocol indicates donors “must be a 7/8 or 8/8 match at HLA-A, -B, -C, and -DRB1”. This is the same sort of donor matching we described five days ago in Do you see what I MHC so perhaps the trial could also record the rs2281389 and rs1800795 genotypes..
Despite the urgent need for a cure for HIV infection, “the risks associated with chemotherapy and radiation, and the relatively low frequency of ccr5
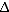 32 homozygous individuals, makes it unlikely that allogeneic HSC transplants using cells from ccr5
32 homozygous individuals, makes it unlikely that allogeneic HSC transplants using cells from ccr5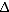 32 homozygous donors will become a widespread treatment option, and has prompted attempts to mimic the genetic knockout”.(PMID 22470838)
32 homozygous donors will become a widespread treatment option, and has prompted attempts to mimic the genetic knockout”.(PMID 22470838)So this type of transplant only directly applies to the fortunately rare cases where a patient has to undergo chemotherapy and radiation to treat their leukemia, enabling a stem cell transplant to repopulate their blood system. It isn’t a practical solution for large numbers of patients. But a ‘SNP transplant’ can be a cure.